
Anyone who starts an investigation of acute myeloid leukemia (AML) soon finds out the complexity of this disease. Although daunting initially, it soon becomes apparent the need for complex classifications for AML subtypes and different mechanisms for formation. AML forms from a wide variety of DNA mutations leading to numerous phenotypic changes in the blood makeup. Early on there were the French-American-British classifications in the 1970s (FAB) but in present day, AML type is being broken down to genetic markers. For the most part, this is due to the advancement of scientific-technical capability. Conversely, being able to clearly define AML by mechanistic function, allows for clinicians to state, with some certainty, treatment and survival options for their patients.
The FAB classification for AML cells is as follows:
M0 undifferentiated
M1 myeloblastic without maturation
M2 myeloblastic with maturation
M3 promyelocytic
M4 myelomonocytic
M5 monocytic
M6 erythroleukemia
M7 megakaryoblastic
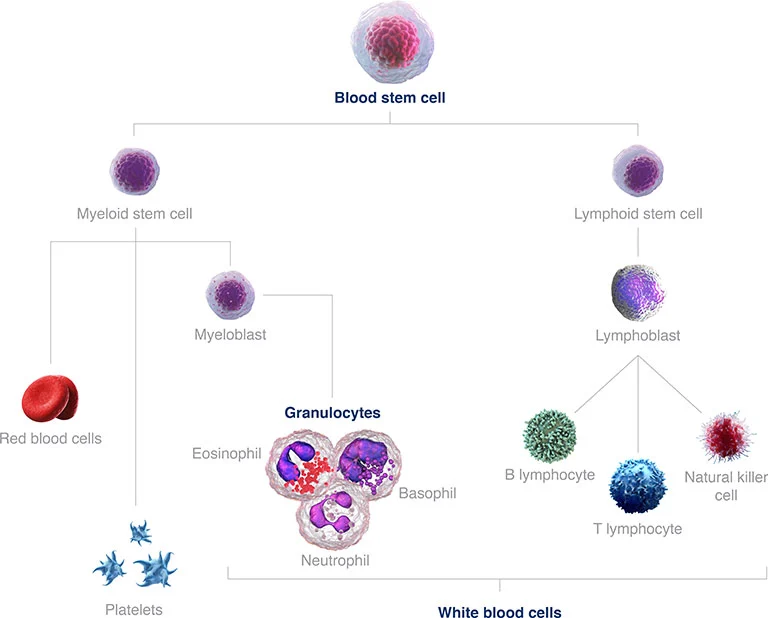
Figure 1: Blood stem cell development with the diversity of mature cells. AML develops from the myeloid stem cell branch.
FAB classification helps to define the differences present in AML, ranging from myeloid to erythroid cells. These are just superficial characteristics though and to be of further assistance to clinicians and scientists, the World Health Organization has broken down the differences between different AML subvariants by gene and chromosome alterations. This allows for greater precision in the study/treatment of AML as well as for clinical discussion with the patients for prognosis.
FMS-like Tyrosine Kinase 3 Receptor and Treatment
In a leukemia condition, there is a subset of immune cells that are growing out of control. For example, in a person with FAB M5 monocytic AML, the monocytic cells can take up approximately 80% of the cells present. Patients have a wide variety of symptoms with the more prominent being bruising, excessive bleeding, or disseminated intervascular coagulopathy. Approximately 30% of leukemia patients have mutant FMS-like tyrosine kinase 3 (FLT3) present. FLT3 plays a role in hematopoietic progenitor cell proliferation and survival. FLT3 mutation is present in most of the FAB classification of AML cells, however, it is measured prominently in the M5 monocytic cells.
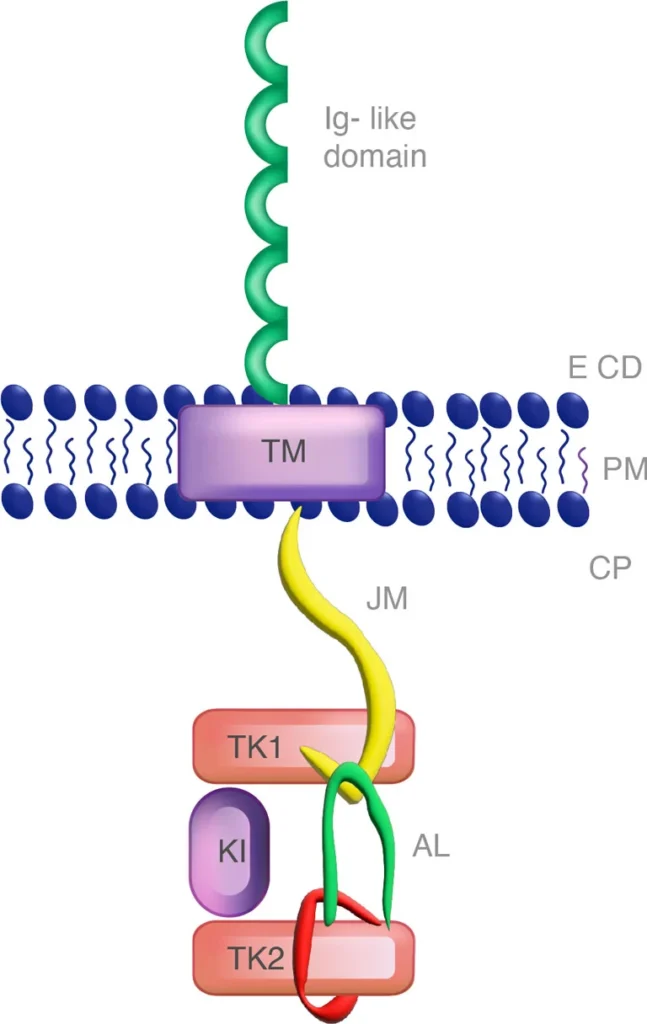
Figure 2: FLT3 monomer with the juxtamembrane (JM) and the tyrosine kinase domain (TKD).
The F691L mutation in FLT3 is critical to the initial evaluation of the patient because it presents resistance to FLT3 inhibitors and a very negative prognosis for the patient. About 1/3 of the FLT3 mutations are within the juxtamembrane duplication (ITD) region while fewer are in the tyrosine kinase domain (TKD) (https://www.xospatahcp.com). Within these regions lie the complexities of FLT3+ AML and the prognosis for the patient. Within the cellular space lies the juxtamembrane region (ITD) and the tyrosine kinase domain (TKD). TKD has the amino acid residues Asp-Phe-Gly (DFG) in the A-loop of FLT3, which is altered upon phosphorylation of the tyrosine residue leading to an active DFG-in conformational status or DFG-out that is inactive (Ke, Y. 2015).
FLT3 inhibitors are in two classifications, Type I and Type II. Type I inhibitors bind near the activation loop or the ATP-binding site. Type II inhibitors bind the FLT3 receptor in the inactive conformation or near the ATP-binding site. Type II inhibitors utilize the hydrophobic nature of the ATP binding site in the DFG-out conformation. Type I inhibitors can be used against mutations in the ITD and TKD region while Type II inhibitors affect only the ITD region. The diagnosis of FLT3 TKD has higher rates of relapse and survival rates (FLT3 TKD mutations have a 53% 5 yr survival rate vs 15% for the FLT3+ ITD)(Smith, C. 2022).
FLT3 Inhibitor Treatment and Phosphorylation Generated Resistance
Gilteritinib is a Type I FLT3+ AML inhibitor. It is a potent treatment for AML patients that have relapsed from cancer. Traditionally, AML patients are initially treated with an intense chemotherapy induction period followed by a consolidation period to produce an AML remission. Relapse occurs in about half the AML patients, unfortunately with a dimmer prognosis usually (Kottaridis, P. 2001). The use of FLT3 inhibitors has developed into a prominent role in FLT3+ AML treatment. FLT3+ AML cells can possess an innate resistance to these FLT3+ AML inhibitors (primary resistance), potentially brought about by the presence of wild-type FLT3 cells (Sato, T. 2011).
Wild-type FLT3 cells present an FLT3/MAPK survival signal that can negate the desired effects of FLT3 inhibitors on the FLT3+ AML cells in vivo (Chen, F. et al. 2016). Other elements such as cytokines and growth factors can initiate MAPK survival pathways in AML cells. Fibroblast growth factor 2 (FGF2) was found to be increased in FLT3-ITD+ AML patients just before relapse and the induction of resistant mutations (Traer, E. 2016). Secondary resistance negates the initial benefits of using an FLT3 inhibitor through additional mutations in FLT3 (“on-target” resistance) and downstream alterations (“off-target” resistance).
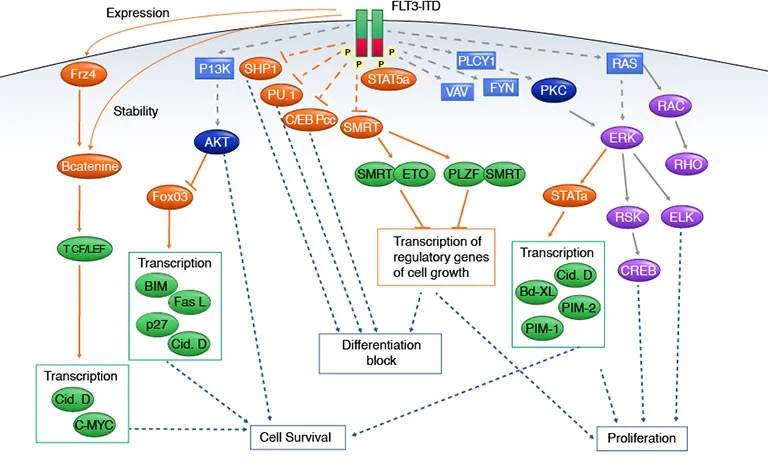
Figure 3: FLT3 signaling pathways.
Secondary resistance to FLT3 inhibitors again leads to more desperate measures in the treatment of the patients. In one study, a few of the resistant patients with no FLT3 mutations, all had alterations to their FLT3/MAPK pathways (McMahon, C. et al. 2019). Among the RAS/MAPK components altered to include the BRAF, NRAS and KRAS, CBL, and PTPN11 mutations. Other pathways that can be altered are PI3K, Akt, and mTOR pathways can be activated by the activation of AXL-1 (a member of the TAM family of TK receptors) (Park, I. 2015). The AXL-1 upregulation is demonstrated to be associated with STAT5 activation in other experiments (Dumas, P. 2019).
Inhibition of specific key pathways can negate the alteration of these pathways and thus reduce the development of secondary resistance (Park, I. 2015). Therapeutics like gilteritinib can provide greater overall survival versus a salvage chemotherapy treatment (Smith, C. 2022). Even with this benefit, RAS/MAPK pathways are still altering the life cycle of the myeloid cells. Even in this second round of therapy due to resistance to the first round, the RAS/MAPK continues to cause resistance to gilteritinib (McMahon, C. 2019; Zhang, H. 2019).
Moving AML Forward
At one point in time, AML was a much simpler disease but that was mainly due to our lack of knowledge. As our science around AML grew and we learned more about this disease, we have learned about the complexities AML entails. Advances in technology have allowed for many of these advances. Technologies such as flow cytometry can decipher numerous cell types and stages of development within a single sample. Flow cytometry can help differentiate AML to the multi-faceted immune cell types involved in the advancement of the disease. Central to treating AML, are the phosphorylation pathways of RAS and MAPK. These pathways or similar phosphorylation events play a key role in the success of the initial treatment and certain follow-up treatments due to relapse.
Measuring changes in these phosphorylation events while treating patients for AML provides evidence of positive or negative turns in prognosis early on. Flow cytometry can also assist with the measure of phosphorylation events in a cell-specific manner. The combination of specific cell treatments and measuring deviations in critical pathways associated with therapy resistance allows for a clear understanding of the direction of AML development in the patient.
References
- Chen, F. Ishikawa, y. Akashi, A. Naoe, T. Kiyoi, H. Co-expression of wild-type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget 2016, 7, 47018-47032.
- Dumas, P. Naudin, C. Martin-Lannerée, S. Izac, B. Casetti, L. Mansier, O. Rousseau, B. Artus, A. Dufossée, M. Giese, A. et al. Hematopoietic niche drives FLT3-ITD acute myeloid leukemia resistance to quizartinib via STAT5-and hypoxia-dependent upregulation of AXL. Haematologica 2019, 104, 2017–2027.
- Ke, Y. Singh, V Coumar, M Hsu, Y. Wang, W. Song, J. Chen, C. Lin, W. Wu, S. Hsu, J.T.A. et al. Homology modeling of DFG-in FMS-like tyrosine kinase 3 (FLT3) and structure-based virtual screening for inhibitor identification. Sci. Rep. 2015, 5, 11702
- Kottaridis, P. Gale, R. Frew, M. et al. The presence of a FLT3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the first cycle of chemotherapy: analysis of 854 patients from the United Kingdom Medical Research Council AML 10 and 12 trials. Blood. 2001;98(6):1752-1759.
- McMahon, C. Ferng, T. Canaani, J. Wang, E. Morrissette, J. Eastburn, D. Pellegrino, M. Durruthy-Durruthy, R. Watt, C. Asthana, S et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019, 9, 1050-1063.
- Park, I. Mundy-Bosse, B. Whitman, S. Zhang, X. Warner, S. Bearss, D. Blum, W. Marcucci, G. Caligiuri, M. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3-targeted therapy in acute myeloid leukemia. Leukemia 2015, 29, 2382–2389.
- Sato, T.; Yang, X.; Knapper, S.; White, P.; Smith, B.D.; Galkin, S.; Small, D.; Burnett, A.; Levis, M.J. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood 2011, 117, 3286–3293
- Smith, C. Levis, M. Perl, A. Hill, J. Rosales, M. Bahceci, E. Molecular profile of FLT3-mutated relapsed/refractory patients with AML in the phase 3 ADMIRAL study of gilteritinib. Blood Advances, 2022, April 12, Vol. 6, Num. 7; 2144-2155.
- Traer, E. Martinez, J. Javidi-Sharifi, N. Agarwal, A. Dunlap, J. English, I. Kovacsovics, T. Tyner, J. Wong, M. Drucker, B.J. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid leukemia. Cancer Res. 2016, 76, 6471-6482.
- Zhang H, Savage S, Schultz AR, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun. 2019;10(1):244