
Why is Viability Staining so important for flow cytometry analysis?
It seems pretty easy, just mix your cells with antibody conjugates, incubate, wash and you are ready to acquire, but actually cell staining for flow cytometry analysis is far from simple, particularly when it comes to the optimal distinguishing of rare cell populations, or in the deciphering of a complex immune response. There are some key considerations to optimizing flow cytometry cell staining and it requires a disciplined approach to understand the impact of certain steps on the staining process. So, how well do you plan and optimize your staining conditions to ensure the highest quality of flow cytometry data?
In this blog, we explore some rules of thumb for Good Staining Practices for flow cytometry
Antibody Selection and Handling
Firstly, the golden rule is always titrate your antibodies- more antibody doesn’t equate to better staining, and the same antibody clone, conjugated to a different fluorophore may titrate quite differently. For some targets, it may also be important to titrate antibodies on your population of interest. Not all cells express markers at the same level, and the only way to ensure that your target cell population is appropriately stained is to titrate and detect the signal from these cells for each marker of interest.
Use flow cytometry analysis software such as FlowJo or FCS Express to analyze your data in order to calculate either your Separation or Staining Indices. At FlowMetric we typically calculate the Separation Index which is used to identify the optimal antibody concentration that provides maximum separation of positive and negative populations and minimizes the spread of the negative population.
A.
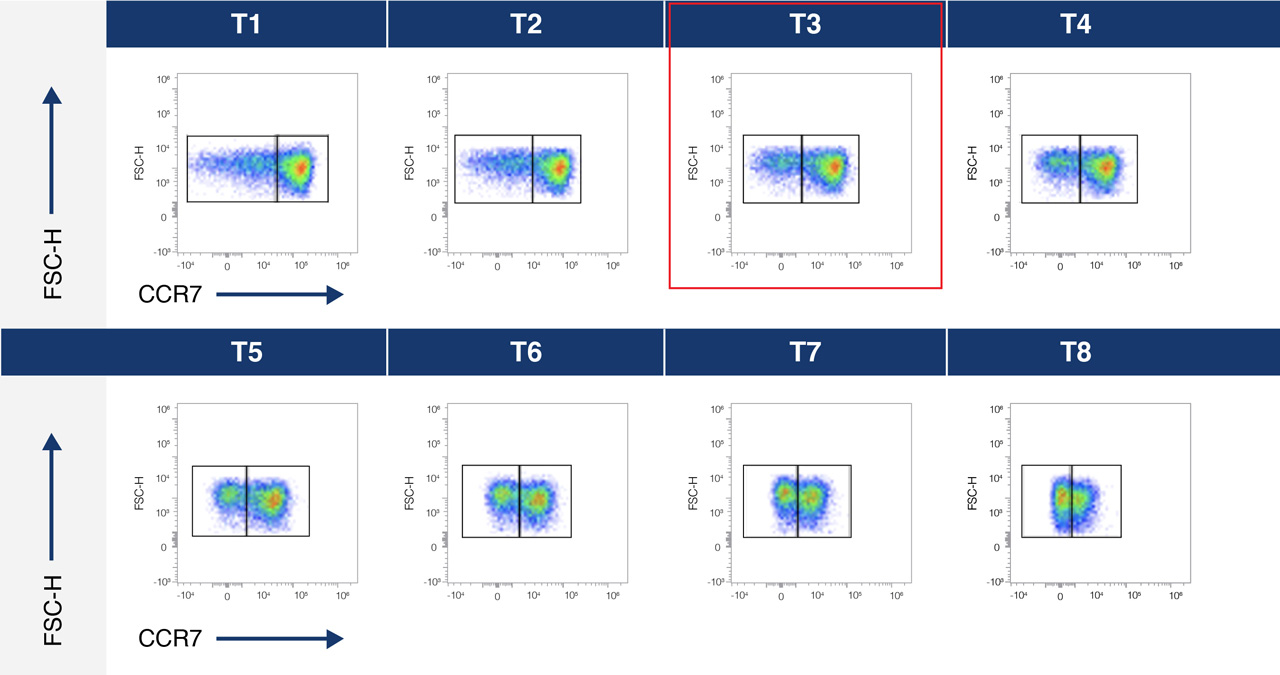
B.
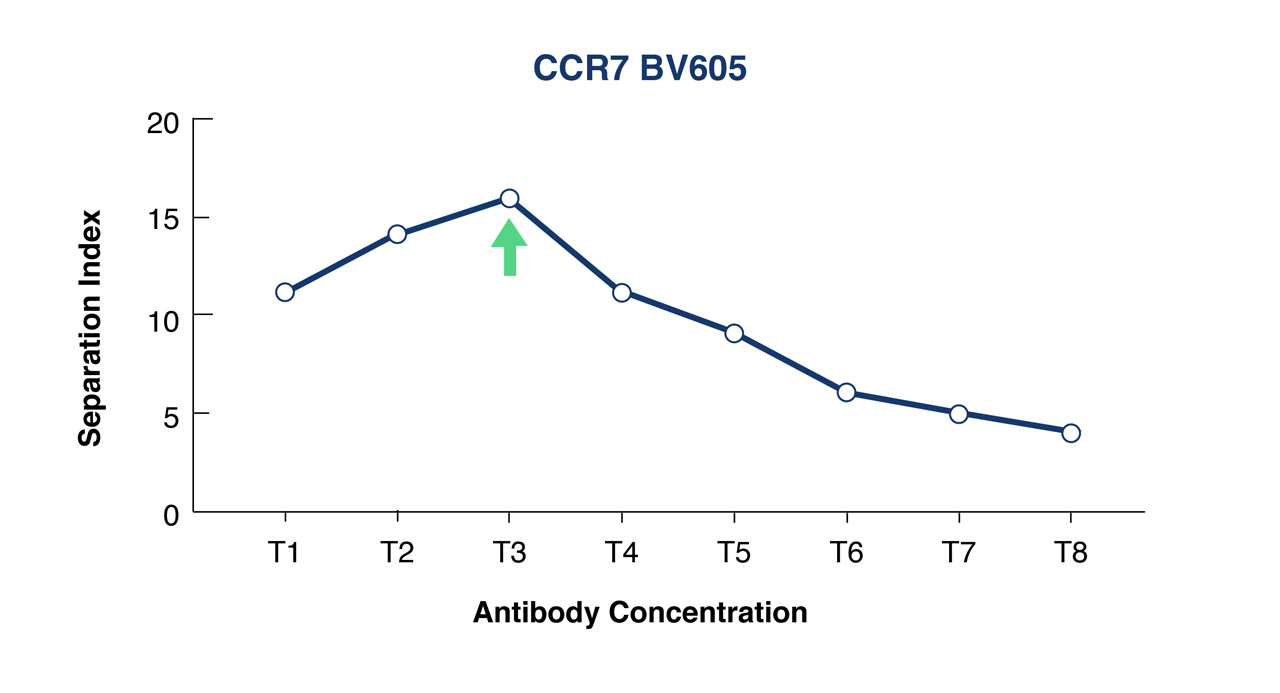
Figure 1A. Titration curve for an anti-CCR7-BV605 conjugate over 8 different concentrations- T1 lowest and T8 highest concentration.
Figure 1B. Separation Index was used to select optimal conjugate concentration for population separation. In this example this is T3.
Try to avoid the use of secondary antibodies for so-called ‘indirect labeling’ as much as possible, but in the event that you do have to utilize secondary detection antibodies there are a couple of key considerations that will help to ensure that the signal detected is real:
- Titrate both the primary and secondary antibodies to ensure optimal signal-to-noise ratio.
- Employ a secondary-antibody only control to determine non-specific binding of this antibody
- Make sure the secondary antibody is selected to only identify the species, class, or isotype of the primary unlabeled antibody.
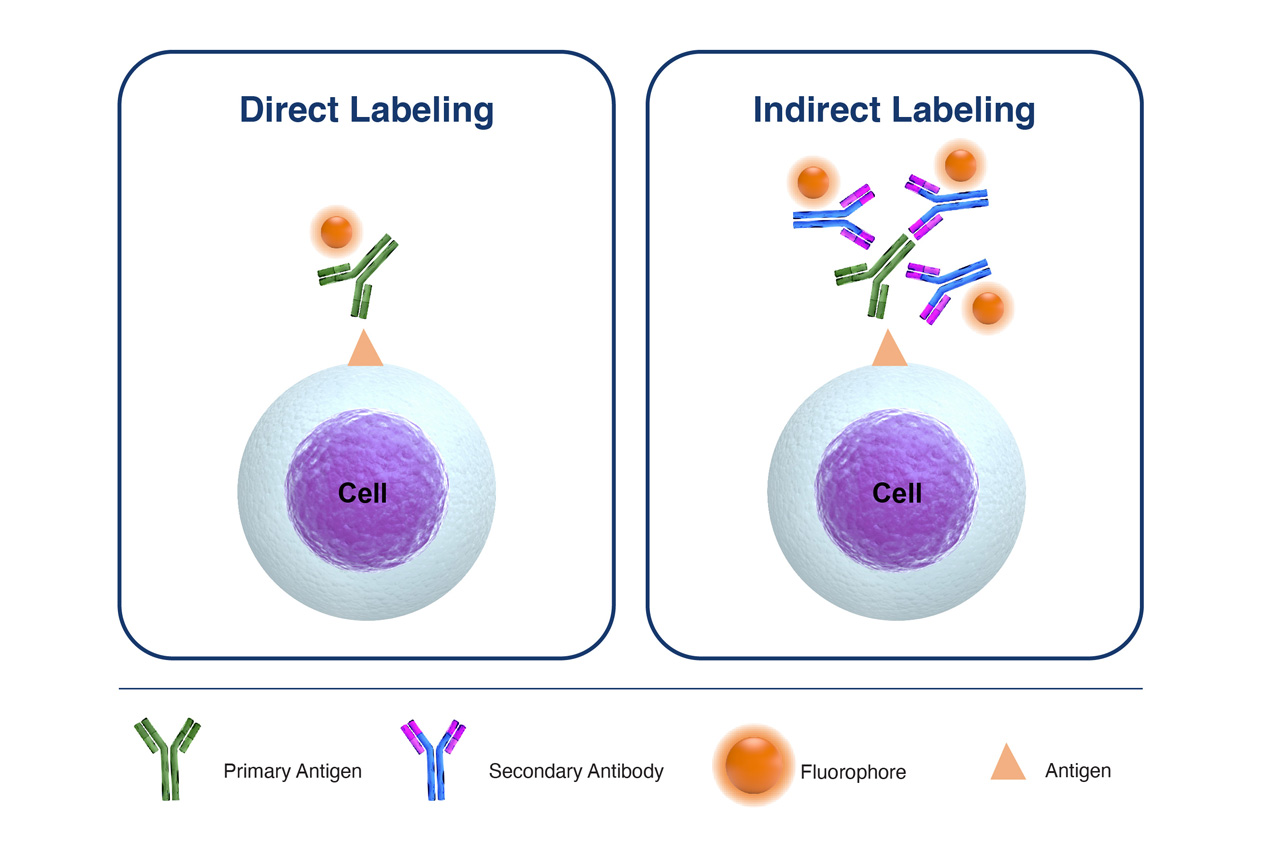
Figure 2. Comparison of Direct Labeling with Primary Fluorophore-Labeled Antibody, versus Indirect Labeling with Unlabeled-Primary Antibody coupled with Fluorophore-Labelled-Secondary Antibody.
The setting up antibody cocktails/master mixes really helps to ensure consistency across all samples within an experiment. The use of antibody cocktails also helps to avoid the pipetting of very small volumes of conjugates- anything below 2µL is very difficult to accurately pipette, and certainly, if you are planning on pipetting less than 1µL it is advisable to prepare a dilution series.
For ease of tracking, always establish an optimal antibody concentration in mg/mL in your protocols. This will help if you have to switch vendors for the same clone or switch to different fluorescent conjugates. By expressing in mg/mL, you have a good starting point for a bridging study as well as instill consistency across staining protocols.
Two of the simplest parameters of cell staining to control for are incubation time and temperature, both of which can have a significant impact on the resulting staining performance. Time for staining can vary anywhere from 15 minutes to overnight, and whereas some antibody conjugates bind optimally at 4˚C, others require room temperature incubation. It is therefore good practice to refer to the manufacturer’s recommended protocols and then keep these as standardized a possible for given targets and clones.
Cell Handling
Apply as much consistency to cell processing and staining across any given experiment as possible. This includes using the same number of cells, and the same antibody concentrations across tubes. When setting up staining controls, it is always better to stain the same number of cells as in the test sample. If cells are limited, then the cell number may be reduced, but then make sure the staining volume is maintained so that final antibody concentration are not changed. If this isn’t possible then compensation beads are used in place of the sample for compensation controls. However, for FMO controls, it is highly recommended to use the same number of cells as in the full panel test sample.
To this end, start out with more cells than you think you will need- you’ll always lose cells along the way, there is no avoiding it -so typically a good starting number is 1×106 cells – the caveat is really what is the frequency of your target cell population in your sample? If this is a relatively abundant cell type, let’s say monocytes, then a starting sample of 5×105 cells is perfectly reasonable, and even after processing, several thousand events would be readily acquired. Even less abundant cell populations, for example, Tregs, which represent about 0.5% of leukocytes, a starting population of 1×106 should yield ~50,000 Tregs; even with a 50% loss in cells during processing, a statistically significant number of Treg events should be acquired. But when it comes to truly rare cell populations, such as circulating tumor cells, that may only represent 0.001% of circulating cells, then starting with 1×106 cells just won’t be enough-so prepare for large sample volumes, high event rates, and long acquisition times. The use of No Lyse/No Wash or Lyse/No Wash reagents during processing will help to eliminate cell loss through washing and centrifugation. Most flow cytometrists aim to acquire 250-500 rare cell events to provide statistical significance, so careful planning and processing might be necessary in order to reach this target.
During the acquisition of rare cell events, it is good practice to keep an eye on the time gate and identify any aberrant occurrences. At FlowMetric, we aim to apply at least two markers to positively label the rare cell population, and another two for which this population is negative. These can be implemented to create a dump gate to eliminate dead/dying cells, cell debris, cell aggregates, and all cell populations with these unwanted markers from the subsequent analysis and focus on the cells stained with our two positive population markers. Adopting this approach can really help ensure optimal staining and support statistically significant detection of rare events.
Staining Issues
When signals are unexpectedly low, firstly check your instrumentation. CS&T or Flow Check beads can be used to ensure the lasers are aligned, and then confirm that the appropriate bandpass filter is in place for the channel being used. If there are still issues, then try re-titrating the antibody conjugate to determine optimal signal-to-noise ratio for the target. Remember that some dyes, particularly tandem dyes are light-sensitive, so using a fresh aliquot of conjugate might help resolve the staining issue.
If the target is intracellular, then the permeabilization step may need to be optimized. Always permeabilize on ice using ice-cold reagents and avoid large fluorophores on conjugates used for intracellular staining. For surface markers, the addition of sodium azide during sample staining will prevent the modulation and internalization of surface antigens that can otherwise result in reduced signal fluorescence. If a panel involves both extracellular and intracellular staining, it is advisable to start with the extracellular staining first, since the fixation/permeabilization process can modify surface antigen availability to antibody binding.
On the flip side, when fluorescent signals are unexpectedly high the most likely causes are excess antibody binding, either because the titer of antibody used is too high, or because of non-specific binding. The inclusion of Fc blocking reagents reduces the binding of antibody conjugates through the Fc receptors on B-cells, monocytes, dendritic cells, and macrophages.
The inclusion of Triton in post-staining wash buffers can also help to reduce non-specific conjugate binding and help optimize signal-to-noise ratios for both surface and intracellular targets.
Unstained Samples
It is really important to include an unstained control sample in order to set thresholds and identify negative gates for each population. This is also helpful in assessing auto-fluorescence, in different cell populations, particularly after fixation.
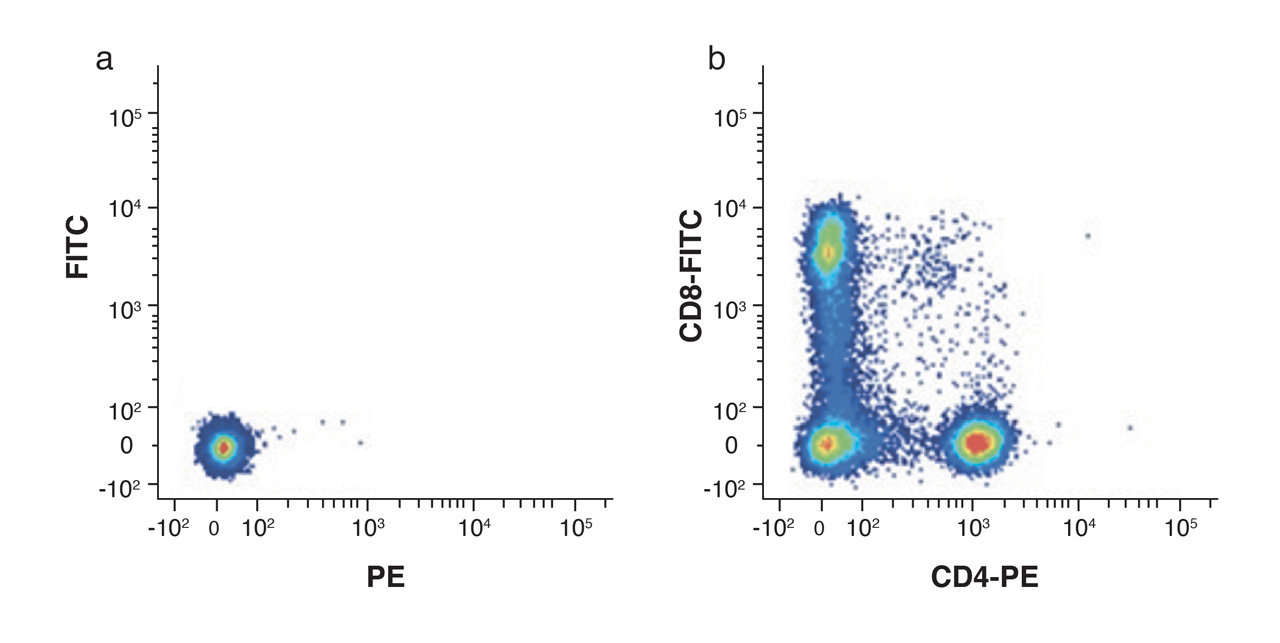
Figure 4. Unstained lymphocytes are used to set the negative population (a) by assessing the background autofluorescence, allowing the positive populations (b) stained with CD8-FITC and CD4-PE to be visualized.
Final Thoughts
Optimal cell staining is a cornerstone of robust flow cytometry analysis and there are several good practices outlined here that can really make a difference. Knowing the biology of your target cell populations and their markers is an important consideration for all experimental design. But careful planning, handling, and consistency in staining practices are central to reproducibility and solid flow cytometry data.